Thank you for visiting my website !
If you did not find the information you are seeking, please Contact me directly.
Dr Benjamin Linard
With the advent of high-throughput DNA sequencing technologies, the research of efficient and proficient bioinformatic algorithms and methodologies became a critical element in many medical, evolutionary and environmental studies.
After studying biochemistry and molecular biology, I specialized in bioinformatics and computer sciences during my Master and PhD Theses. Since then, I continuously developed my research interests and skills towards the fields of environmental and evolutionary sciences.
My past projects spanned over a large area of environmental/medical applications with at their core the development of new bioinformatic algorithms, software, databases and analysis workflows.
Below is a short list of keywords summarizing the main subjects I studied during the past years:
- ALGORITHMS
classification, phylogenetics, sequence alignment ... - SOFTWARE
Python, Java, C++, web frameworks (Django), SQL, DBMS platforms, ... - DEVOPS
cluster computing, web services, reproducibility, continuous integration, ...
- EVOLUTIONARY GENOMICS :
genome assembly, genome annotation, orthology inference, biological networks, ... - ECOLOGY :
trophic interactions, microbial communities, symbiosis, ... - BIODIVERSITY :
taxonomy, systematics, aquatic/soil biodiversity, ...
Short CV
Current affiliation

Past affiliations







Education
- 2012 : Phd Bioinformatics, University of Strasbourg
- 2007 : Master Bioinformatics & Structural Biology, University of Strasbourg
- 2006 : License (bachelor) in Biology & Computer Science
- 2005 : Technological diploma (DUT) Biochemistry and food industry
Short Bio
- Since 2019 : research officer in Spygen R&D department
(Hosted in LIRMM, Montpellier, France)
Development of a bioinformatic platform support for the Vigilife Consortium
- optimization and software development for eDNA analysis
- web frameworks for eDNA analyses submission
- digitisation of the R&D processes
- establishment of public / private partnerships & co-developments.
-
2018 : Research assistant. Univ Montpellier, LIRMM / ISEM / AGAP, France.
LabEx Project Orthologap: development of a new algorithm to classify sequences into families of orthologous genes.
- phylo-k-mer based supervised classification.
- orthology and paralogy.
- genomic sequence annotations.
- 2016 : Research assistant, CNRS, LIRMM, Montpellier, France.
European Consortium Virogenesis.
Development of an alignment-free phylogenetic placement method.
- development of the phylo-k-mers idea, algorithms for their computation.
- phylogenetic placement for taxonomic identification.
- applications in virome analysis.
- viral genome recombination detection.
- 2013 : Research assistant, Natural History Museum of London, United Kingdom.
Project Biodiversity Initiative :
"Breaking the taxonomic barrier"
- Development of the methods of mitochondrial metagenomics and metagenomic skimming.
- Applications in systematics, phylogenetics and biodiversity discovery.
- biodiversity of Coleoptera and soil arthropods.
- metagenomics for gut content and trophic interactions.
- 2009 : PhD Student, University of Strasbourg, IGBMC, Strasbourg, France.
Projects Evolhupro and OrthoInspector.
Development of algorithms and software for comparative genomics, genome annotation
and multi-scale omics data integration and visualisation.
- bipartite graph-based clustering for orthology and paralogy inference.
- multi-scaled omics data integration (genome, proteome, biological networks).
- omics data representation and machine learning.
Research
Main topics
Axis 1 : « phylo-k-mers » for the classification of biological sequences
I study the analytical breakthrough offered by the "phylo-k-mers",
an abbreviation for "phylogenetically informed substrings of length k".
Phylo-k-mer should not be confounded with classical k-mers extracted
from real sequences (those correspond to the state of the sequences at the terminals).
From a set of known
reference sequences and a tree that represent their relationships, phylo-k-mers can be computed on
the basis of models of evolution and algorithms of ancestral sequence reconstruction.
Then, for a particular tree branch, it is possible to generate
the set of k-mer that diverged from it and their associated probabilities.
There is (#states^k) possible k-mer for each reference alignment position and tree branch.
Extended to all alignment positions and tree branches, computing phylo-k-mers rapidly becomes a challenging task,
and leads to many open questions :
how to choose the divergence points ? which algorithm should I use to explore this huge sequence space ?
is the approach scalable to large tree and alignments ? how does it behave with sequencing error and
alignment mistakes that would not fit the model of evolution ?
Still, as a first step it is possible to learn a large sequence space, the set of phylo-k-mers,
from a relatively small reference dataset. Then, in a second step,
classifiers can be developed to exploit phylo-k-mers in many applications, from taxonomic or functional
classification of biological sequences,
to the detection of evolutionary phenomena, for instance genome recombination.
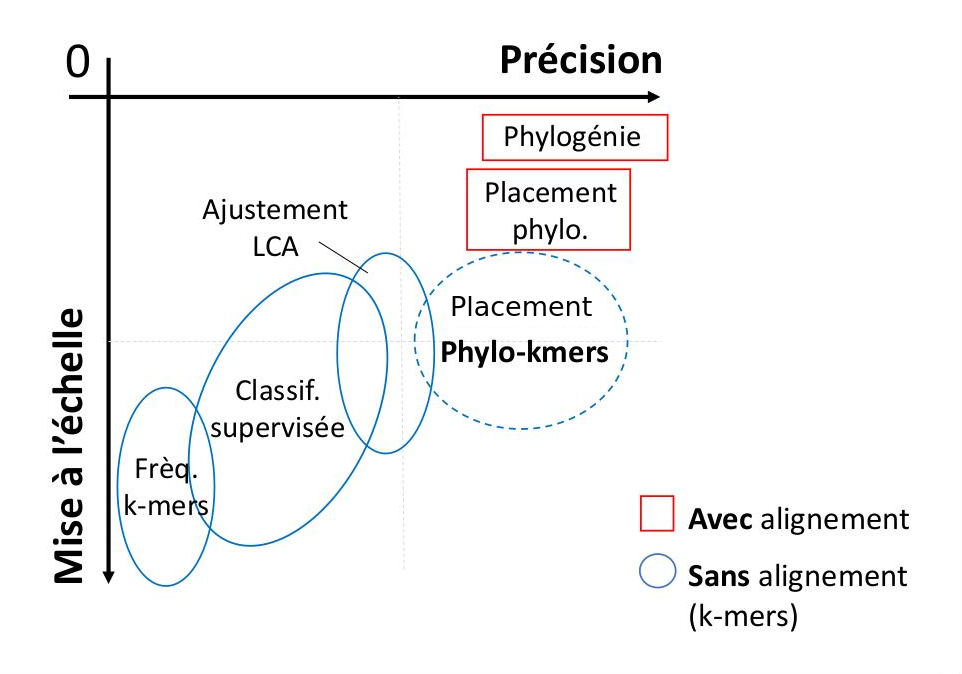 With this approach I developed RAPPAS, the first alignment-free
algorithm of phylogenetic placement, (Linard et al, 2019) a task of sequence classification exploited
in the critical process of taxonomic identification of metagenomic and metabarcoding sequences.
With this approach I developed RAPPAS, the first alignment-free
algorithm of phylogenetic placement, (Linard et al, 2019) a task of sequence classification exploited
in the critical process of taxonomic identification of metagenomic and metabarcoding sequences.
I was also involved in the development of SHERPAS (Scholz zt al, 2020), a tool
designed for viral genome recombination detection. In both projects
the classification phase demonstrated an acceleration of several orders of magnitude when compared to other
available tools. This opened new possibilities
for routine sequence-based taxonomic identification and diagnostics.
In collaboration with colleagues from classical phylogenetics, and
exploring the theoretical relationships between the phylo-k-mer approach and
machine learning techniques, I recently focused on applications in genome functional annotation.
Phylogenetic Placement occurred as a thriving research field over the past years,
and in this context I got interested in means of benchmarking this classification process.
This question lead me to develop PEWO,
a platform for benchmarking placement tools. With this work, I try to alleviate
the critical questions that arise between developers and end-users of phylogenetic placements :
which placement algorithm and implementation best fits a particular input dataset ? Is their
accuracy / computational resources trade-off adapted to a particular experimental design.
Axis 2 :
orthology inference for genome annotation, comparative genomics and phylogenomics
Today genome sequencing is a tool accessible to many research laboratories and new genomes are
sequenced at an increasing rate. The evolutionary relationships
of Orthology links homologous genes issued from a speciation event.
Because ortholog genes tend to be more similar in their biological function
than paralogs, inferring accurate orthology predictions is a fundamental step in
genome annotation, gene function analysis and phylogenomics.
This inference is however a difficult task. Incomplete genomes, domain loss and gains, gene loss,
horizontal transfers, and fast-paced divergence between genes and gene families are some phenomenon explaining
why new inference approach are continuously developed by the research community.
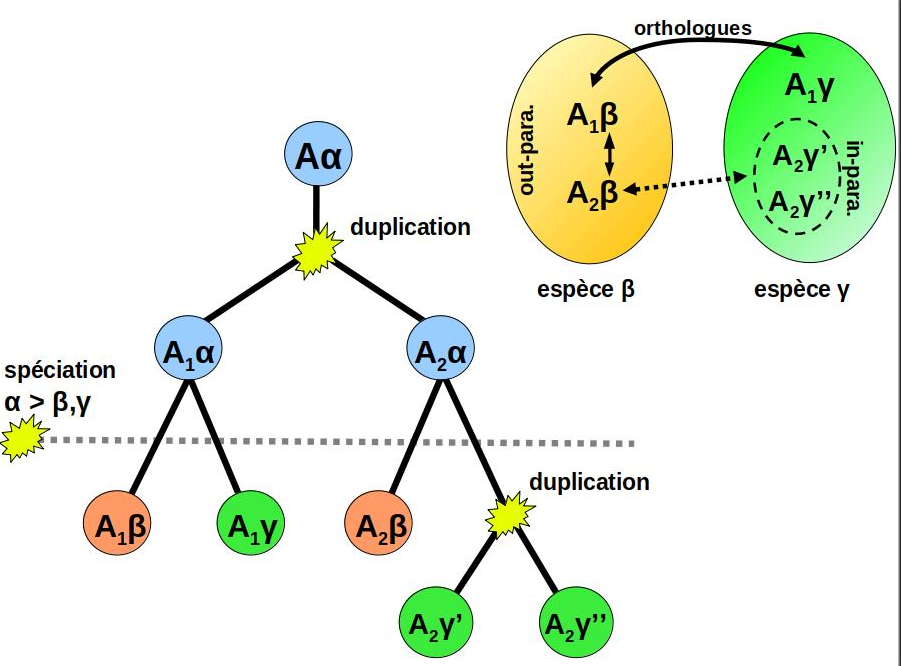 Graph-based orthology inference
is generally considered as an interesting alternative to the more computationally-demanding
phylogeny-based approaches (eg: species and gene tree inference + reconciliation).
During my Phd, I developed Orthoinspector, an algorithm based on bi-partite graphs and design to infer
orthologs relationships between the proteomes of two genomes.
By extension, compiling every pairwise genome comparison for a set of genomes allow deducing orthologs
between hundreds to thousands of complete genomes (Linard et al, 2011).
Eight years and thousands of new complete genomes later,
the Orthoinspector database
still remains one of the largest online resource for pre-computed orthologs
(Never et al, 2018).
Graph-based orthology inference
is generally considered as an interesting alternative to the more computationally-demanding
phylogeny-based approaches (eg: species and gene tree inference + reconciliation).
During my Phd, I developed Orthoinspector, an algorithm based on bi-partite graphs and design to infer
orthologs relationships between the proteomes of two genomes.
By extension, compiling every pairwise genome comparison for a set of genomes allow deducing orthologs
between hundreds to thousands of complete genomes (Linard et al, 2011).
Eight years and thousands of new complete genomes later,
the Orthoinspector database
still remains one of the largest online resource for pre-computed orthologs
(Never et al, 2018).
The challenges of orthology lead me to join the
Quest for Ortholog consortium. This workgroup aims
to provide standards for the orthology community and maintains a benchmarking platform that evaluates
the quality of new predictions.
More recently, I started to explore a link between the phylo-k-mer approach (axis 1) and one of the usage
application of pre-computed orthology relationships: high-throughput and accurate classification of
coding sequences into known gene families.
Axis 3 : Pipelines relevant to evolutionary & environmental applications
Beyond applied algorithms and software implementation, my research focuses on the methodological challenges
encountered by the rapid development of environmental genomics.
The success of an approach based on environmental DNA (eDNA), often means
tight integration between every methodological steps, from sampling, DNA extraction,
DNA sequencing, to bioinformatic analysis and data integration.
Through international projects and consortiums, I specialized into the analysis of biological sequence sampled in
different types of environments (soil, water, gut contents...). Not only am I interested in improving the generation
of the DNA data itself, but I also aim for data integration with other features (ex: environmental or geographical parameters)
and novel applications of data mining.
From a field point of view, I studied an alternative approach of eDNA sequencing,
labeled "metagenome skimming" (Linard et al, 2016),
as well as its more selective derivative, the approach of "mitochondrial mitogenomics" (Crampton-platt et al, 2016).
These approaches are an alternative to metabarcoding when long taxonomic markers (> several kb), are seeked for
poorly known ecosystems. As longer sequences generally means more phylogenetic signal, these approaches
are an interesting basis for species-rich phylogenetic reconstruction, a critical step not only in systematics,
but also for biodiversity studies where the completeness of reference database is essential.
These project made me participate in a large panel of thematics, including NGS protocols optimisation,
arthropod systematics, biodiversity indices and the extraction of trophic interactions
as well as microbial symbiotic interactions from gut contents.
More recently, I focused on large-scale bioinformatic analysis of metabarcoding data and issues related to
analysis reproducibility and scalability.
All together, tight interactions with researchers in Ecology, Evolution, Biodiversity and Conservation give
me a vision encompassing every step of a research project based on eDNA. I aim to convert this expertise
into the development of expert bioinformatics tools and pipelines that are adjusted to those applications.
Publications
See the DEDICATED PAGE
for a complete list of my scientific publications.
Other options :
Software & databases
Repositories
 |
Phylo42 : Phylo-k-mer dev. centralization |
github |
Software
| Orthology inference |
webpage | v1.0 - v3.0 Linard et al, 2012 Linard et al, 2015 Nevers et al, 2019 |
|
| Bioanalysis pipelines : - Mitochondrial Metagenomics - Metagenome Skimming | QC, Assembly, Annotations |
Linard et al, 2015 Linard et al, 2016 Andujar et al, 2019 |
|
| RAPPAS : Alignment-free phylogenetic placement |
github | Linard et al, 2018 | |
| . | SHERPAS : Alignment-free recombination detection |
github | Scholz et al, 2020 |
 |
PEWO : Phylogenetic placement benchmarking |
github | Linard et al, 2020 |
| Coming soon | CLAPAS : Phylo-k-mer based classification of coding sequences |
. | :) |
Databases
 |
Pre-computed orthologs for thousands of complete genomes |
website | v1.0 - v3.0 Linard et al, 2012 Linard et al, 2015 Nevers et al, 2019 |
| KD4V : (Knowledge Discoveryfor Missense Variant) |
webpage | Dao et al, 2012 | |
| Visual exploration of gene family evolutionary histories |
Currently down |
Linard et al, 2012 |
Other contributions
 |
Regular contributor |
github | . |
| Taxonomy Java Tools | Java library | github | Linard et al, 2018 |
 |
Dataset contributions to the QFO consortium |
website | Altenhoff et al,2016 Forslund et al,2018 Linard et al, 2021 |
|
|
Metabarcoding analysis plateform |
spygen.com vigilife.org |
Tutoring
Professional Tutoring
CNRS formations (since 2017)
- "Molecular Phylogeny" & "Advanced Phylogeny" -
New modules introducing to the algorithms and applications of phylogenetic placement of environemental data.
- "NGS Data Analysis" -
New module focusing on OTU clustering and taxonomic classification.
Bioinformatics workshops
- 2013-2016 : NHM, London -
New 2 days workshops for the Phd Program introducing to sequence analysis and metagenomics.
- 2010-2012 : IGMBC, France -
Yearly intervention in the internal bioinformatics workshop.
Students supervision
- Co-supervision of 1 Phd thesis.
- phylo-k-mer computation and phylogenetic placement. (director Eric Rivals) - Regular supervision of Licence (Bachelor) and Master students. Currently 10 student (2021). Some projects lead to co-authorship. (ex: Orthoinspector v2)
University lectures
So far, I had the chance to give lecture and practical courses for 3 different universities
(Strasbourg ; Imperial College London ; Montpellier), for more than 380 hours of effective teachings.
Depending on the subjects, those were given for Computer Science, Ecology/Environment,
or Health/Agronomy or Earth Sciences departments. For simplification,
I regroup these interventions under the following thematics:
Computer sciences

- Algorithms in bioinformatics (data representation, trees, graph, clustering, phylogeny...)
- Introduction to programming : Python, Shell, Java ...
- Introduction to Unix environments & shell scripting
- Models for object programming (UML, software design)
- Advanced Java Programming (GUI, RDBMS, networking, threading ...)
- Data sciences : multivariate data analysis & visualisation, python, R, jupyter ...
- Introduction to Devops : Gitflow, continuous integration, containers (Docker)
- Bases of population dynamics: competition, selection, models, practical courses in web lab.
- Practical courses for the analysis of NGS / metagenomic data
- Introduction to sequence databases and sequence analysis (data extraction, alignment, phylogeny)
- Introduction to eDNA approaches and applications
- Bioinformatic methods for genome annotation (de novo & reference-based)
- Methodological approaches for comparative genomics
- Introduction to Transcriptomics / Proteomics
- Functional annotation : orthology, gene ontology, biological networks
Public outreach
In a world were scientific facts are regularly challenged,
I am deeply convinced that scientific outreach and vulgarisation are critical subjects.
When time permits, I am participating to activities related to science vulgarisation in schools and
in public events. Below is a non-exhaustive list of events in which I was or continue to be involved
either to create new activities or give public talks.
- Openlab :
A program lead by the University of Strasbourg, in which Phd Students go to regional High School and give a practical course introducing DNA molecular analysis and genetics as a Police enquiry scenario. - Fêtes de la science :
Regular participation to this French national day dedicated to science outreach.
I mostly propose activities introducing what are bioinformatics. In particular, I created some mini-games introducing the discrepancies between species traits (ecology, morphology...) and DNA similarity (evolutionary relationships).
In the same context and in partnership with the Académie de Montpellier, I intervene into priority area schools to introduce kids to the field of science, what is the university and what is the job of a researcher. My key point being that these are not inaccessible jobs.
-
 Université Tiers Temps Montpelier :
Université Tiers Temps Montpelier :
An open university service of the university of Montpellier teaching a large array of scientific, literary, law subjects to adults. I gave general talks related to bioinformatics and environmental DNA.
- Science Uncovered :
An event organized by the Natural History Museum of London in which scientists have a direct contact with the public. With my former colleagues, we were leading recreationnal activities of DNA extraction and talks related to environmental DNA and biodiversity exploration.

Contact
For professional enquiries :
| University lectures (Montpellier) |
||
| Spygen | website | |
| LIRMM | website |
For other inquiries : gmail